I want to perform some MD including Li+ ions. Specifically I am interested in Li-TFSI (aka Lithium bis(trifluoromethanesulfonyl)imide).
I tested both the ATB repository and LigParGen and neither can provide Li parameters.
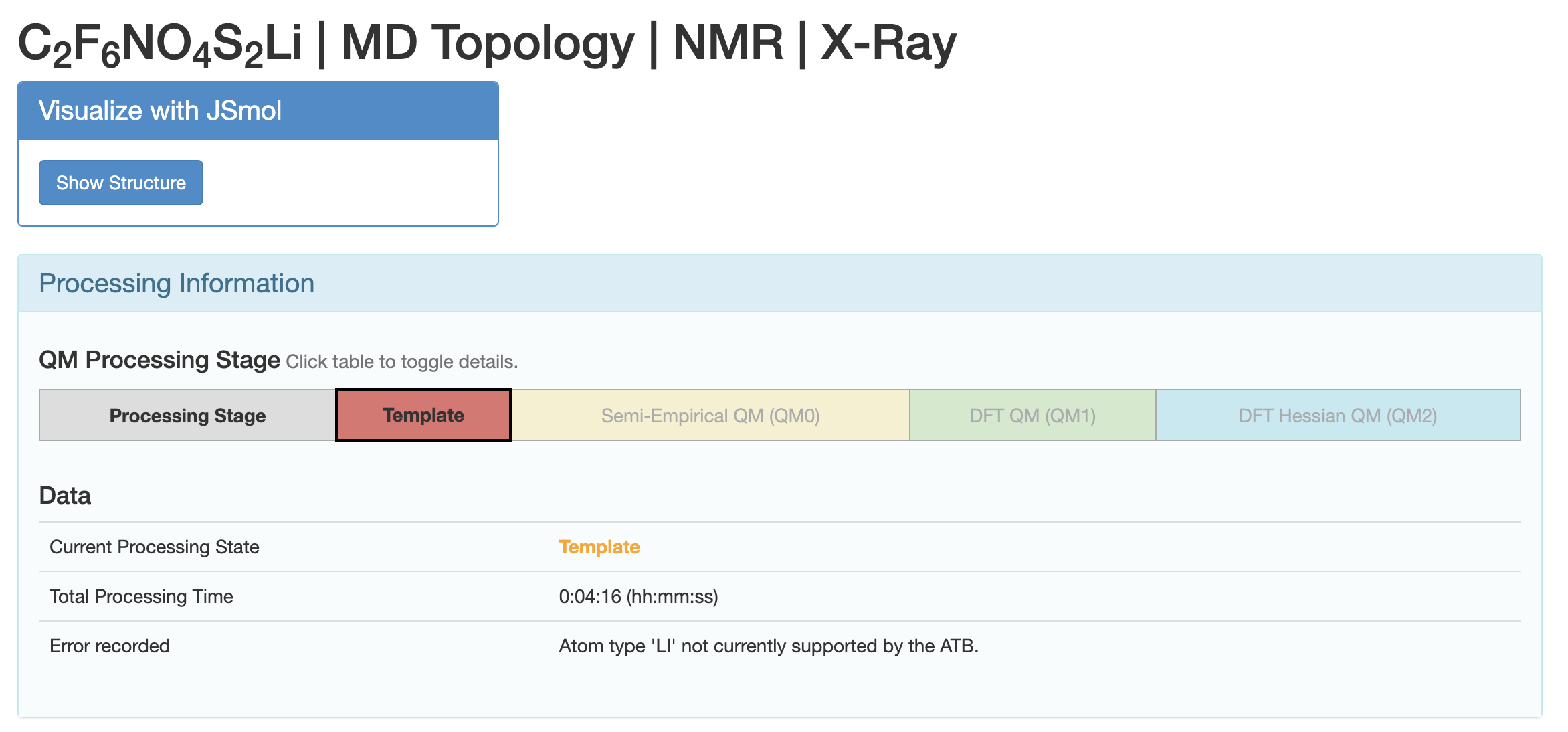
The calculation 1273328 on ATB fails since Atom type 'LI' not currently supported by the ATB.
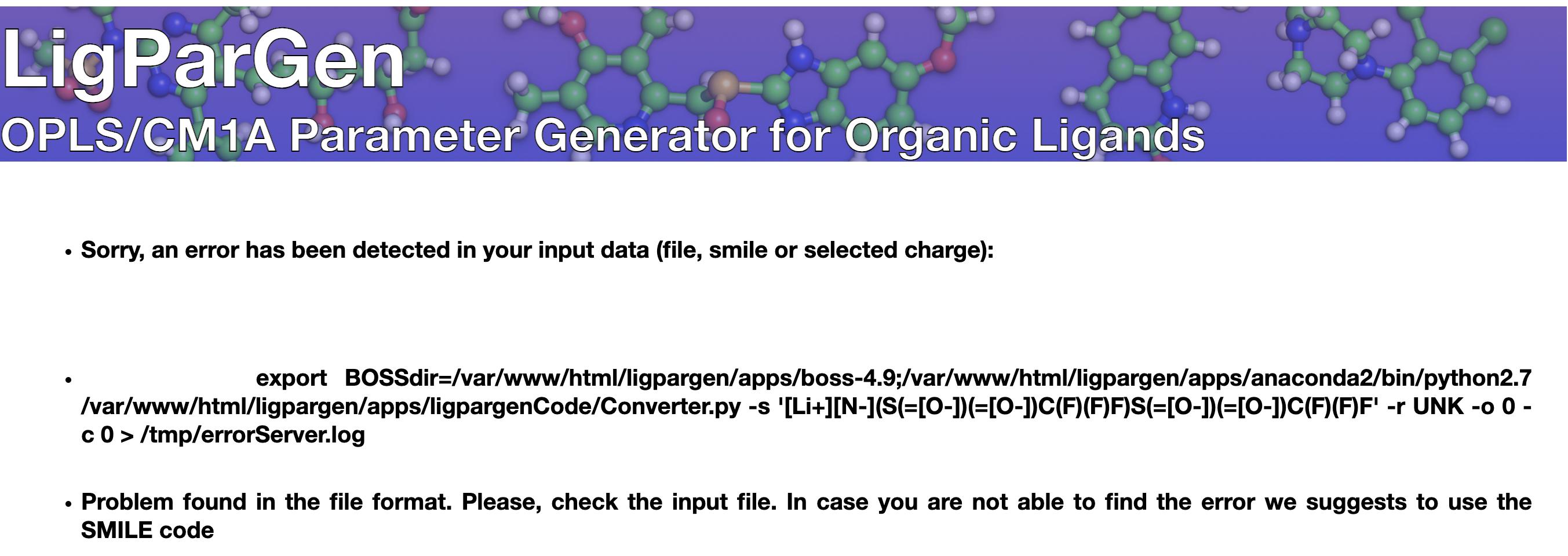
 On LigParGen there is no Lithium allowed
On LigParGen there is no Lithium allowed export BOSSdir=/var/www/html/ligpargen/apps/boss-4.9;/var/www/html/ligpargen/apps/anaconda2/bin/python2.7 /var/www/html/ligpargen/apps/ligpargenCode/Converter.py -s '[Li]' -r UNK -o 0 -c 0 > /tmp/errorServer.log
I tried two smiles as FC(F)(F)S(=O)(=O)N([Li])S(=O)(=O)C(F)(F)F and [Li+][N-](S(=[O-])(=[O-])C(F)(F)F)S(=[O-])(=[O-])C(F)(F)F

I tried also to search the Li related parameters into OPLS from Yale or in abmer but I couldn't find much. In amber I tried to run antechamber on my Li-TFSI.pdb input file, obtaining this:
Warning: Unusual element (Li) for atom (ID: 16, Name: Li).
/Users/marcodigennaro/miniconda3/envs/lammps/bin/wrapped_progs/antechamber: Fatal Error!
GAFF does not have sufficient parameters for molecules having unusual
elements (those other than H,C,N,O,S,P and halogens).
Question: Do you know any Li parametrization to add to these force fields?
Many thanks
LIin any of the input files perhaps? $\endgroup$