I have ORCA 5.0.4, OpenMPI 4.1.1 and SLURM in UBUNTU 22 in a single PC with 16 threads.
I have setup this pc and, when I run orca alone, openmpi works. But, when I submit orca through slurm, the calculation fails. In this image I can show what is happening:
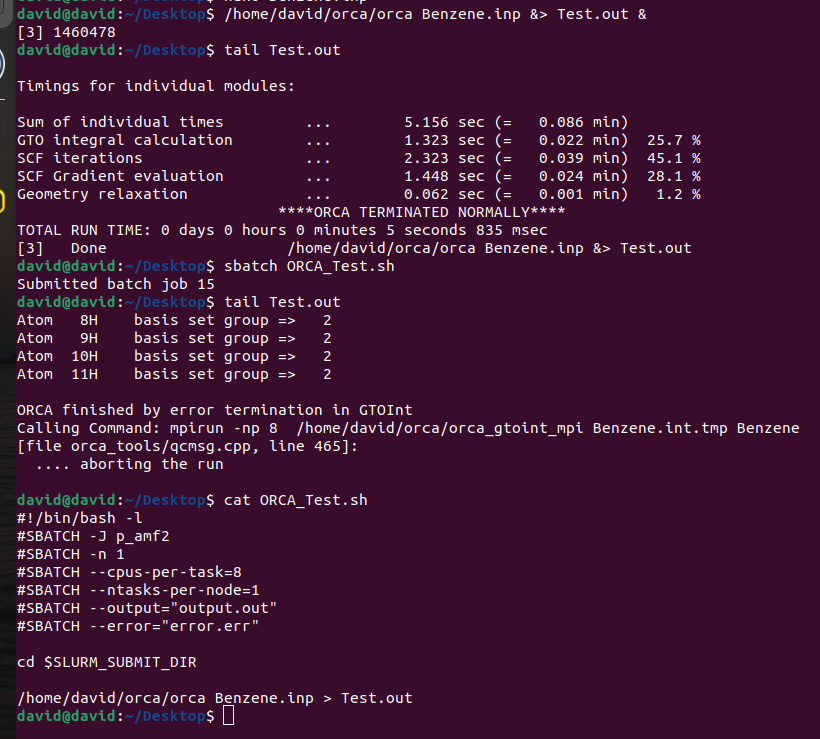
It seems that SLURM does not recognize OpenMPI
How can I resolve it?
Files:
Orca.sh
#!/bin/bash -l
#SBATCH -J p_amf2
#SBATCH -n 1
#SBATCH --cpus-per-task=8
#SBATCH --ntasks-per-node=1
#SBATCH --output="output.out"
#SBATCH --error="error.err"
#SBATCH --mem=1000
export LD_LIBRARY_PATH="$HOME/orca:$LD_LIBRARY_PATH"
export LD_LIBRARY_PATH="/usr/local/lib/openmpi/:$LD_LIBRARY_PATH"
export MPIROOT="/usr/local/bin/:$MPIROOT"
export OMP_PROC_BIND=true OMP_PLACES=sockets
export OMPI_MCA_mpi_yield_when_idle=1
cd $SLURM_SUBMIT_DIR
/home/david/orca/orca Benzene.inp > Test.out
ORCA input:
# Benzene RHF Opt Calculation
#%pal nprocs 8 end
! RHF TightSCF PModel
! opt
# Molecule specification
* xyz 0 1
C 0.000000000000 1.398696930758 0.000000000000
C 0.000000000000 -1.398696930758 0.000000000000
C 1.211265339156 0.699329968382 0.000000000000
C 1.211265339156 -0.699329968382 0.000000000000
C -1.211265339156 0.699329968382 0.000000000000
C -1.211265339156 -0.699329968382 0.000000000000
H 0.000000000000 2.491406946734 0.000000000000
H 0.000000000000 -2.491406946734 0.000000000000
H 2.157597486829 1.245660462400 0.000000000000
H 2.157597486829 -1.245660462400 0.000000000000
H -2.157597486829 1.245660462400 0.000000000000
H -2.157597486829 -1.245660462400 0.000000000000
*
slurm.conf
# slurm.conf file generated by configurator.html.
# Put this file on all nodes of your cluster.
# See the slurm.conf man page for more information.
#
ClusterName=localcluster
SlurmctldHost=localhost
MpiDefault=none
ProctrackType=proctrack/linuxproc
ReturnToService=2
SlurmctldPidFile=/var/run/slurmctld.pid
SlurmctldPort=6817
SlurmdPidFile=/var/run/slurmd.pid
SlurmdPort=6818
SlurmdSpoolDir=/var/lib/slurm/slurmd
SlurmUser=slurm
StateSaveLocation=/var/lib/slurm/slurmctld
SwitchType=switch/none
TaskPlugin=task/none
#
# TIMERS
InactiveLimit=0
KillWait=30
MinJobAge=300
SlurmctldTimeout=120
SlurmdTimeout=300
Waittime=0
# SCHEDULING
SchedulerType=sched/backfill
SelectType=select/cons_tres
SelectTypeParameters=CR_Core
#
#AccountingStoragePort=
AccountingStorageType=accounting_storage/none
JobCompType=jobcomp/none
JobAcctGatherFrequency=30
JobAcctGatherType=jobacct_gather/none
SlurmctldDebug=info
SlurmctldLogFile=/var/log/slurm/slurmctld.log
SlurmdDebug=info
SlurmdLogFile=/var/log/slurm/slurmd.log
#
# COMPUTE NODES
NodeName=localhost CPUs=24 RealMemory=31772 State=UNKNOWN
PartitionName=LocalQ Nodes=ALL Default=YES MaxTime=INFINITE State=UP
--ntasks-per-node=8be more appopriate? given that MPI creates multiple tasks $\endgroup$--cpus-per-task=1is also needed, I implicitly assumed the OP would try this, but without your comment they didn't $\endgroup$