I'm interested in calculating elastic constants for 2D (monolayer) using VASP and I'm looking for guidance on the necessary steps. Could anyone here provide some insight into how to go about this process effectively? Any tips or resources would be greatly appreciated.
$\begingroup$
$\endgroup$
Add a comment
|
1 Answer
$\begingroup$
 $\endgroup$
$\endgroup$
3
Theoretical Background:
Elastic constants can be calcualted in one of two approaches. First is via the stress-strain relationship, and the second is by strain-energy relationship.
First approach:
In solids, $\sigma$ or stress response to $\varepsilon$ strain (external loading) follows the Hooke’s law which is simplified in the Voigt notation: $$\tag{1}\sigma_i = \sum_{j=1}^{6}c_{ij}\varepsilon_j$$
In this relation, both stress and strain are vectors with 6 components each. While $C_{ij}$ is a tensor written as 6x6 matrix known as the second order elastic stiffness tensor, which can be determined based on the first order derivative of the stress-strain curves as you can see in the figure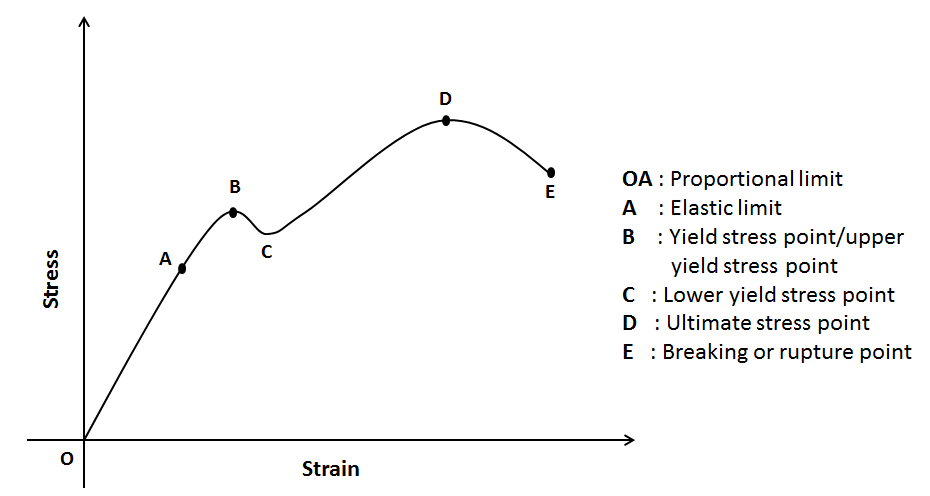
Second approach:
The second approach is based on the variation in energy when applying small strains (you have to add that manually) to the equilibrium lattice. In this method, the elastic stiffness tensor is derived from the second-order derivative of the total energies versus strain in harmonic approximation: $$\Delta E(V,\left\{ \varepsilon_i \right\})= E(V,\left\{ \varepsilon_i \right\}) - E(V_0,0) = \frac{V_0}{2} \sum_{j=1}^{6}c_{ij}\varepsilon_j \varepsilon_i\tag{2}$$
The first two terms are total energies of pristine and distorted lattices with volumes $V_0$ and $V$.
How To:
In VASP and to my knowledge, only stress-strain method is applied and it is accessible when you set IBRION=6 and ISIF>=3, according to VASP Docs: The elastic tensor is determined by performing six finite distortions of the lattice and deriving the elastic constants from the strain-stress relationship. The elastic tensor is calculated both, for rigid ions, as well, as allowing for relaxation of the ions.
For the other approach, you can use automated tools to save you the time as you need to add the distortion. One example is to use vaspkit. Detailed explanation is available from the documnetation and I will mention that in here just in case that link might not be active in the future.
Prepare the fully-relaxed POSCAR file including the standard conventional cell of your structure. Run vaspkit with task of 102 to generate KPOINTS with fine k-mesh. Prepare following INCAR:
Global Parameters
ISTART = 0
LREAL = F
PREC = High
LWAVE = F
LCHARG = F
ADDGRID= .TRUE.
Electronic Relaxation
ISMEAR = 0
SIGMA = 0.05
NELM = 40
NELMIN = 4
EDIFF = 1E-08
Ionic Relaxation
NELMIN = 6
NSW = 100
IBRION = 2
ISIF = 2 (Must be 2)
EDIFFG = -1E-02
Prepare VPKIT.in file and set the value of first line to 1 (1 means pre-processing)
1 ! 1 for pre-processing; 2 for post-processing
2D ! 2D for two-dimentional, 3D for bulk
7 ! number of strain case
-0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 ! Strain range
Run vaspkit-201, upon which you will create the required folder in the working directory, each of which corresponds to the strain you have introduced, next you will batch perform DFT calucaltions using VASP code in each of those folders. Lastly, Modify the value of the first line in VPKIT.in file to 2 (2 means post-processing); and run vaspkit-201 again and you will get the calculated elastic constants in the output.
Finally
The first approach of stress-strain method require higher computational precise to achieve the same accuracy as energy-strain method. Nevertheless, the former requires much smaller set of distortions than the latter. There exist other tools out there that you might want to check as well such as ELasTool and Elastool
answered Feb 29 at 14:43
-
$\begingroup$ @Jaafer Mehrez While preparing VPKIT.in file can we change strain range? $\endgroup$ Commented Mar 1 at 8:04
-
$\begingroup$ @FarahShehzadi, This is just a sample file, you can choose the range according to your study and requirments $\endgroup$ Commented Mar 1 at 8:26
-