The question is related essentially to solve the Kohn-Sham equation with an atomic-like basis set. In fact, different implementations of DFT are distinguished mainly by their basis set and how they orthogonalize themselves to the core levels. In particular, the choice of basis set forms the core of any electronic structure method.
Dedepending on the choice of basis set and how to orthogonalize, four methods are proposed as shown in the following figure (Nuclei are shown as dots):
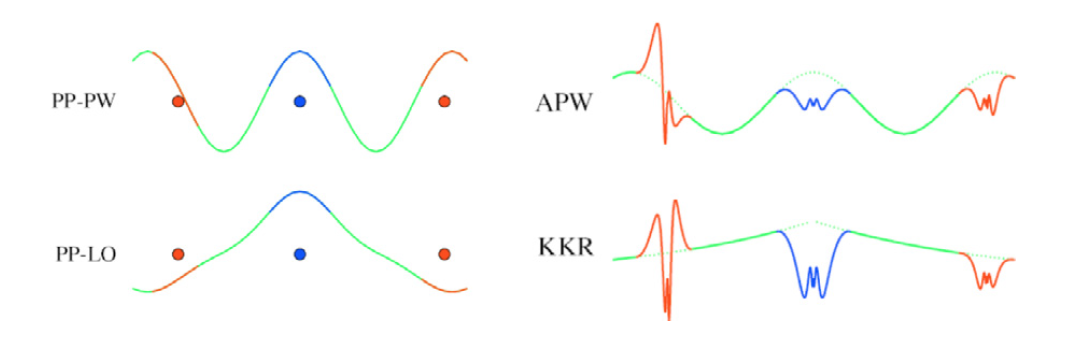
- The all-electron methods APW and KKR on the right substitute (augment)
the envelope function (green) with numerical solutions of partial waves inside
augmentation spheres (blue and red). Parts inside augmentation spheres are
called ‘‘partial waves’’.
- The two figures on the left use a pseudopotential
allowing their envelope functions to be smooth, with no augmentation needed.
A pseudopotential’s radius corresponds to a characteristic augmentation radius.
- The top two figures use plane waves for envelope functions; the bottom two
use atom-centered local basis sets.
The example implementation for these methods:
- PP-PW: VASP
- APW: Wien2K
- PP-LO: Gaussian
- KKR: Questaal (This is what you are looking for!)
For a more detailed comparison between these methods, you can take a look at the implementation paper of Questaal.
When are atomic-orbital-basis (rather than plane-wave) methods appropriate in periodic DFT?
If you want to combine NEGF with DFT to calculate the transport properties of the device, you should use atomic-orbital-basis, as Questaal adopted.