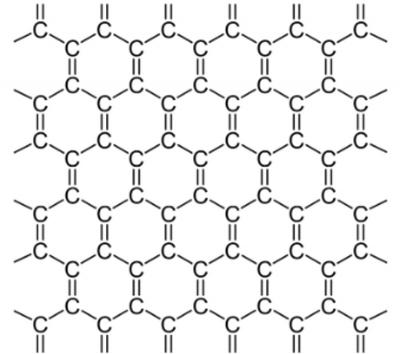
When creating a Graphene layer in VMD using the Nanotube Builder it only generates hexagons with single bonds because of this errors occur when creating the topology for gromacs software using,
gmx x2top -f graphene.pdb -o system.top
Here are the errors observed in gromacs output
Can not find forcefield for atom C-4 with 3 bonds
Can not find forcefield for atom C-6 with 3 bonds
Can not find forcefield for atom C-7 with 3 bonds
Can not find forcefield for atom C-8 with 3 bonds
Please not that the border line C atoms will assign H atoms this can be done with another software which is not the major issue its missing double bonds thus carbon is missing its full 4 bonds.
Can anyone help me with this issue or please suggest any other software that can do this.