You linked to my answer about parameterizing force fields. For reference, this isn't my area of expertise, although a colleague's group does this frequently.
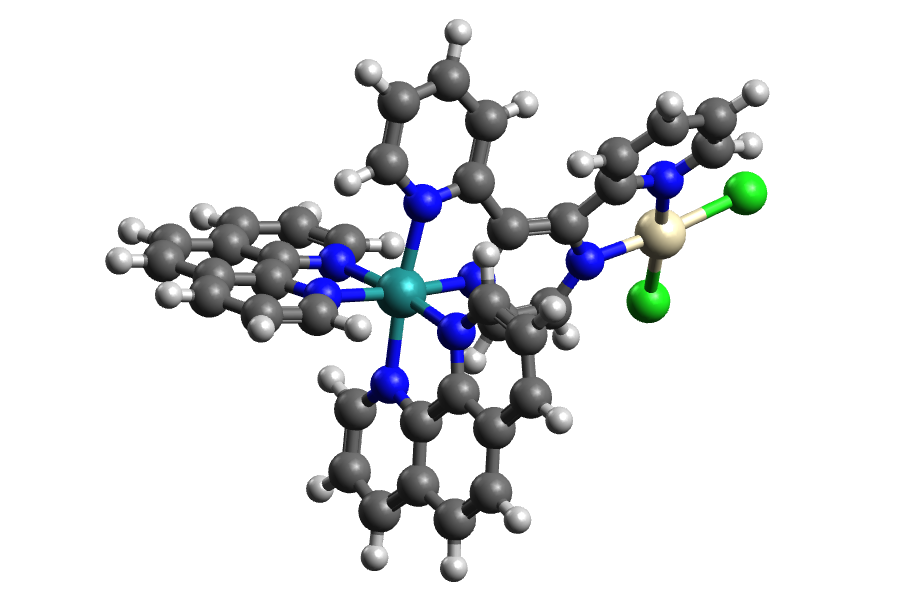
My understanding of your question is that you want to get the non-bonded interactions between the Ru and Pt atoms in the larger complex.
What you describe is going to be tricky, even with automated force field methods like I described on Chem.SE.
In your case, you don't have neutral metal ions - you have transition metals with a surrounding ligand sphere.
Ordinarily, you'd pick the relevant elements, maybe start from the initial distance between them, scan closer and scan farther until you see a full minima. (You want at least parts of the repulsive and attractive regions to fit the potential energy curve.)
For metals, it's more complicated. There are a few approaches.
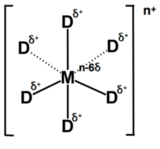
I'd probably go with the "dummy atom" method - you surround the metal ions (Ru and Pt in your case) with partially charged dummy atoms, so the net charge is identical, but the ion is properly surrounded with the right coordination environment and has some charge delocalization.
A fairly readable article is "Force Field Independent Metal Parameters Using a Nonbonded Dummy Model".
So you'd add some dummy atoms to the Ru and Pt at the positions relevant in the complex (e.g., remove the carbon atoms and hydrogen around both, then change the nitrogen atoms around the Ru to dummy (Du) and the two nitrogen and two chlorine atoms around the Pt atom.
Then map the curve .. again starting near the distance in the complex, going shorter and longer until you've mapped out the LJ curve.
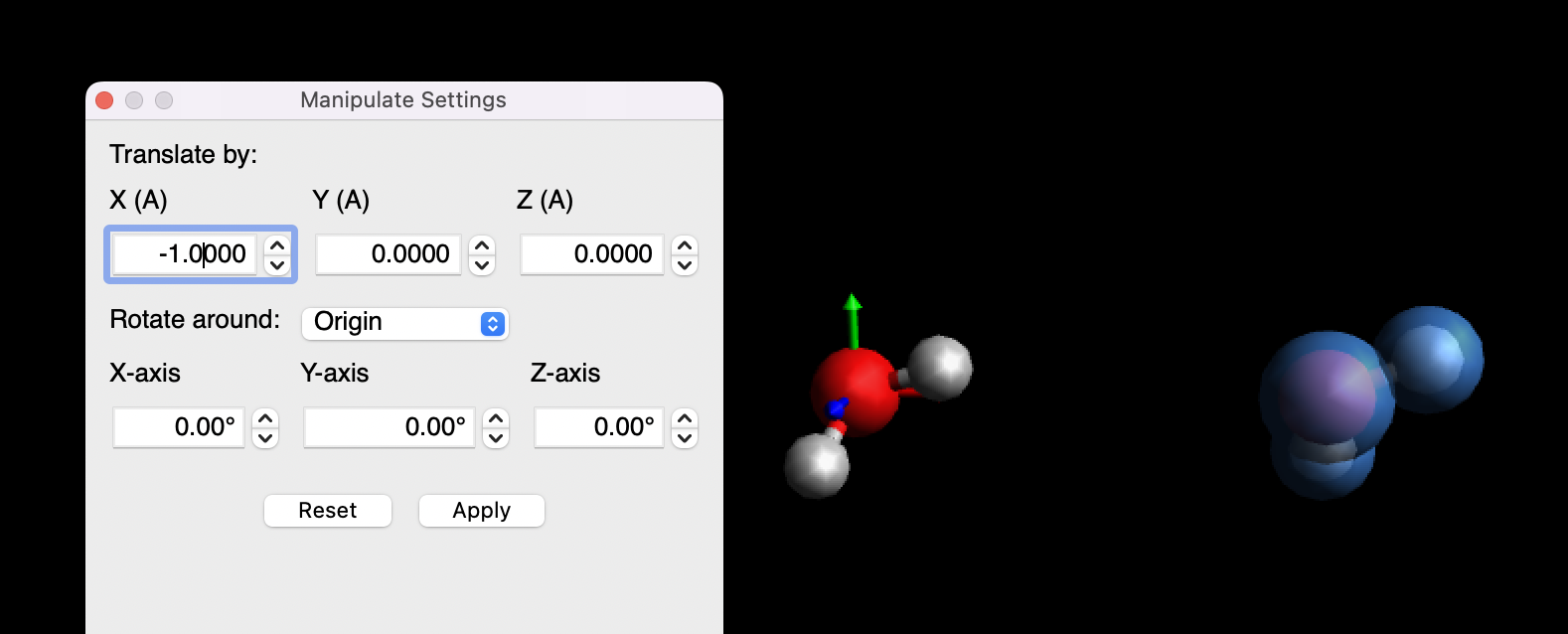
Since it looks like you're using Avogadro, you can use the "Align" tool, click on the Ru atom to make that the origin, then the Pt atom to make that along one of the axes (e.g, x-axis). Then you can select the Pt+dummy "molecule" and use the manipulate tool to translate it forward and backwards.
Merz also has a series of articles talking about the development of parameters for water models (TIP3P, etc.). These are probably less relevant to your needs, but discuss some of the challenges.
I'm probably leaving off relevant papers.