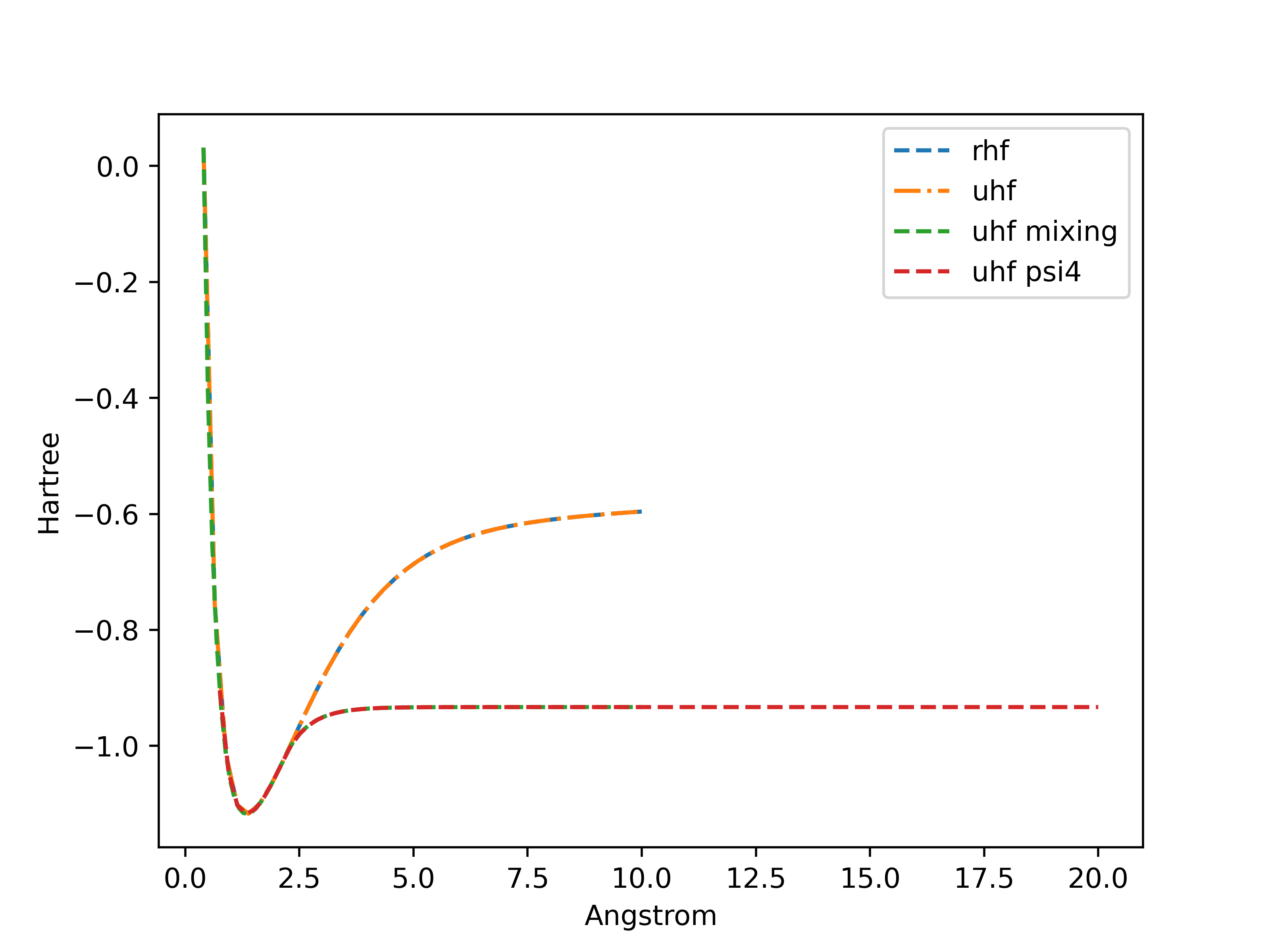
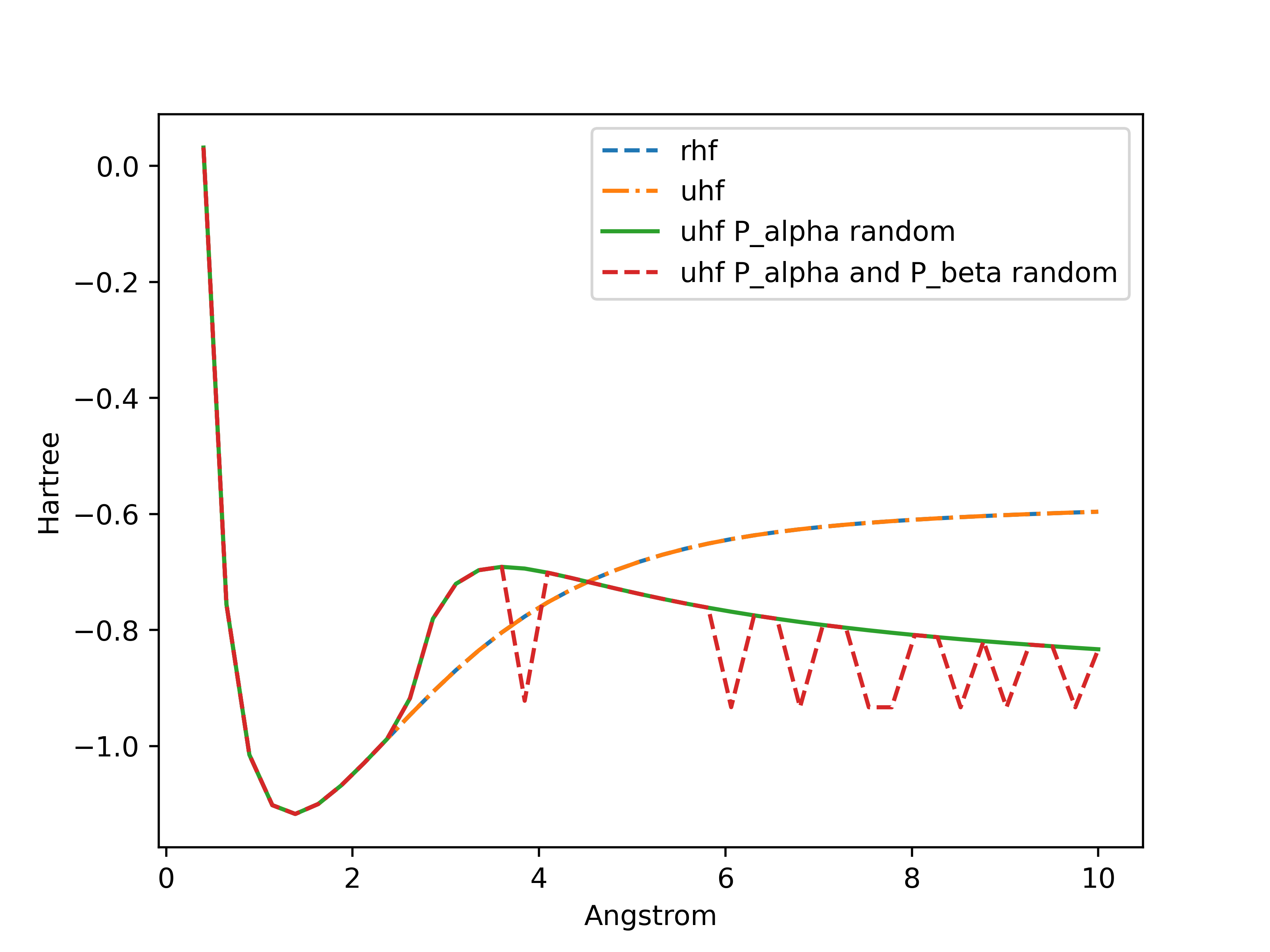
I have some results about the energy of H2 as a function of the bond length using Restricted HF (RHF) and Unrestricted HF (UHF) methods. With a zero-initialization of the density matrix I have the same results which is expected. But When I use a random initialization (Uniform on [0,1]) for both density matrices, the scf loop converges to weird values. And when I only initialize one of the density matrices I have what I suppose to be the UHF solution.
How are density matrices initialized in Unrestricted calculations (UHF, Unrestricted Kohn-Sham)? Is there any schemes to have a more robust algorithm?
legend:
- uhf = UHF with zero initialization of both matrices
- uhf P_alpha random = P_alpha is initialized with uniform distribution on [0, 1] and P_beta as zeros
- uhf P_alpha and P_beta random = both matrices are initialized with uniform distribution
Edit:
I forgot the Coulomb interaction between the alpha and beta orbitals and I used rotation mentionned in Tyberius comment.
As initial guess I used the HOMO and LUMO orbitals of H2:
$$C_{\alpha} = C_{\beta} = \frac{1}{\sqrt{2}}\begin{pmatrix} 1 & 1\\ -1 & 1 \end{pmatrix}$$
Then I rotated the $\alpha$ orbitals, with $\theta = \frac{\pi}{2}$ : $$ C^{new}_{\alpha} = \begin{pmatrix} cos(\theta) & -sin(\theta)\\ sin(\theta) & cos(\theta) \end{pmatrix}C_{\alpha}$$ Here are the results: