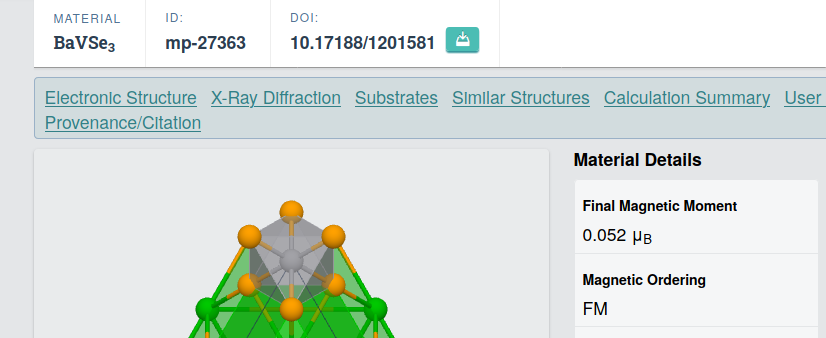
I'm quite new to spin-polarized DFT and have been trying to study BaVSe_{3} which is ferromagnetic (with a Final Magnetic Moment of 0.052 Bohr Mag) as per the Materials Project repository [picture 1]. The Vanadium carries the moment as per this paper. But when a spin-polarized SCF was executed [as per the input file 1], the total magnetization was obtained as 1.95 Bohr Mag/cell. The two values are quite different so am I doing something wrong here? Or are these values different things?
I'm using the primitive cell CIF file obtained from the Materials Project repo [Cif file] and have converted it directly into the Quantum ESPRESSO input file using cif2cell
Picture 1
input file 1
&CONTROL
calculation = 'scf'
outdir = './outdir'
verbosity = 'low'
tprnfor = .true.
tstress = .true.
pseudo_dir = '.'
/
&SYSTEM
ibrav = 0
A = 7.02663
nat = 10
ntyp = 3
ecutwfc = 55
ecutrho = 650
nspin = 2
starting_magnetization(1) = 1
starting_magnetization(2) = 0
starting_magnetization(3) = 0
occupations = 'smearing'
smearing = 'mv'
degauss = 0.005d0
/
&ELECTRONS
conv_thr = 1e-8
mixing_beta = 0.7d0
/
CELL_PARAMETERS {alat}
0.866025403784439 -0.500000000000000 0.000000000000000
0.000000000000000 1.000000000000000 0.000000000000000
0.000000000000000 0.000000000000000 0.857028191323579
ATOMIC_SPECIES
V 50.94150 V.pbe-spnl-kjpaw_psl.1.0.0.UPF
Ba 137.32700 Ba.pbe-spn-kjpaw_psl.1.0.0.UPF
Se 78.96000 Se.pbe-dn-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
Ba 0.333333333333333 0.666666666666667 0.250000000000000
Ba 0.666666666666667 0.333333333333333 0.750000000000000
Se 0.833430000000000 0.666666666666667 0.250000000000000
Se 0.833236666666667 0.166570000000000 0.250000000000000
Se 0.166570000000000 0.833236666666667 0.750000000000000
Se 0.333333333333333 0.166763333333333 0.250000000000000
Se 0.166763333333333 0.333333333333333 0.750000000000000
Se 0.666666666666667 0.833430000000000 0.750000000000000
V 0.000000000000000 0.000000000000000 0.000000000000000
V 0.000000000000000 0.000000000000000 0.500000000000000
K_POINTS {automatic}
17 17 17 0 0 0
Picture 2

CIF file
# CIF file created by FINDSYM, version 7.1
data_findsym-output
_audit_creation_method FINDSYM
_cod_database_code None
_cell_length_a 7.0266300000
_cell_length_b 7.0266300000
_cell_length_c 6.0220200000
_cell_angle_alpha 90.0000000000
_cell_angle_beta 90.0000000000
_cell_angle_gamma 120.0000000000
_cell_volume 257.4939303923
_symmetry_space_group_name_H-M "P 63/m 2/m 2/c"
_symmetry_Int_Tables_number 194
_space_group.reference_setting '194:-P 6c 2c'
_space_group.transform_Pp_abc a,b,c;0,0,0
loop_
_space_group_symop_id
_space_group_symop_operation_xyz
1 x,y,z
2 x-y,x,z+1/2
3 -y,x-y,z
4 -x,-y,z+1/2
5 -x+y,-x,z
6 y,-x+y,z+1/2
7 x-y,-y,-z
8 x,x-y,-z+1/2
9 y,x,-z
10 -x+y,y,-z+1/2
11 -x,-x+y,-z
12 -y,-x,-z+1/2
13 -x,-y,-z
14 -x+y,-x,-z+1/2
15 y,-x+y,-z
16 x,y,-z+1/2
17 x-y,x,-z
18 -y,x-y,-z+1/2
19 -x+y,y,z
20 -x,-x+y,z+1/2
21 -y,-x,z
22 x-y,-y,z+1/2
23 x,x-y,z
24 y,x,z+1/2
loop_
_atom_site_label
_atom_site_type_symbol
_atom_site_symmetry_multiplicity
_atom_site_Wyckoff_label
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_occupancy
_atom_site_fract_symmform
Ba1 Ba 2 c 0.33333 0.66667 0.25000 1.00000 0,0,0
V1 V 2 a 0.00000 0.00000 0.00000 1.00000 0,0,0
Se1 Se 6 h 0.83343 0.66686 0.25000 1.00000 Dx,2Dx,0
# end of cif