TL;DR If bonds are broken in creating the lower surface of the slab, then passivation is probably a good idea.
Let's start with a little bit of context:
Slab calculations in computational materials science are often (but not always) designed to study the properties of a surface of a macroscopic crystal.
In these cases, an accurate model of physical reality would involve a slab of macroscopic thickness.
Unless you are open to using Green-function techniques for modeling a semi-infinite slab (which may not be readily available in standard codes), this is not going to be computationally tractable.
It is also not necessary: you just need your slab to be thick enough to converge the surface property of interest to the accuracy you desire.
While the number of layers of the slab is your most straightforward convergence parameter, increasing the number of layers can substantially increase the number of atoms in your calculation, i.e. be very expensive.
This opens the door for computational tricks to achieve convergence of surface properties at lower numbers of layers - e.g. making the bottom of the slab more "bulk-like" by fixing the lattice constants of some bottom layers to their known bulk value(s) and/or by passivating unwanted surface states at the bottom of the slab through the addition of (pseudo-)hydrogens (which only adds 1 electron per atom => cheap).
This brings us to your question:
When do I need to passivate a slab's bottom side with pseudohydrogen?
One obvious answer is that you don't really need to - you can always1 achieve convergence by increasing the thickness of your slab.
So, when would passivation be useful to improve the accuracy of the calculation?
I'm sorry to say that I don't have a rigorous answer to this question2 but it seems reasonable to think that passivation will be useful, if bonds are broken in the process of creating the lower surface of the slab.
Of course this rule depends on the notion of a bond.
It certainly applies not only to the breaking of covalent bonds (which would result in the "dangling bonds" you mention): the Au(111) surface is an example of a metal surface with a Shockley surface state, which decays exponentially towards the bulk.
The figure below from the Ph.D. thesis of Nora Gonzalez Lakunza shows the difference between a bare slab (left) and a hydrogen-passivated slab (right).
In the bare slab, the surface states from the top and bottom of the slab overlap, forming bonding and antibonding linear combinations, which give rise to an artificial band splitting in the surface band structure that can affect how the surface interacts with adsorbed molecules (details in Appendix I.b).
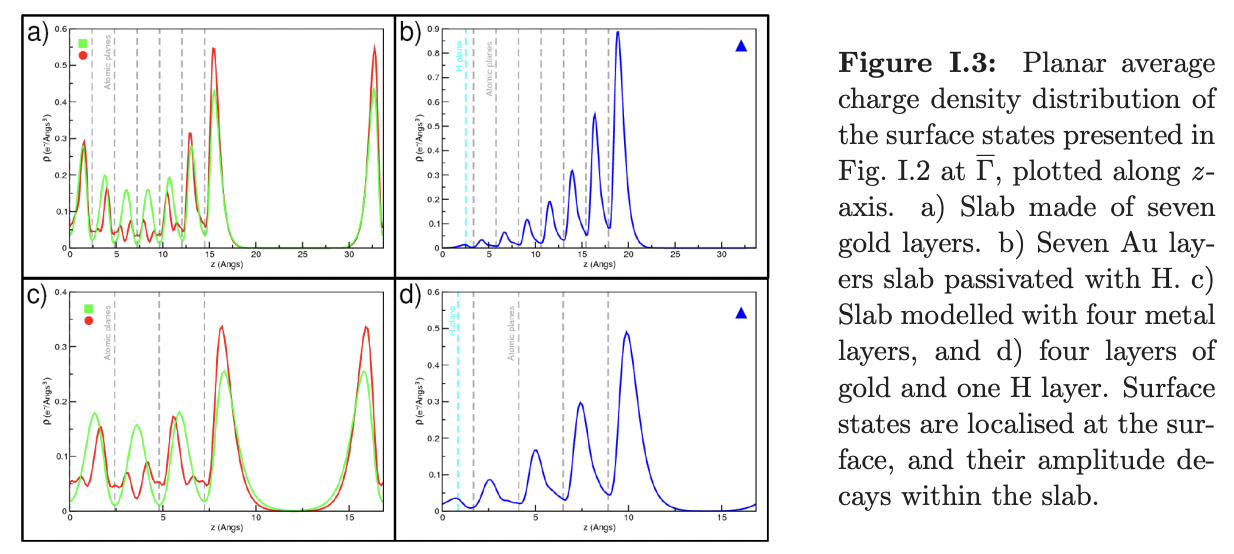
As shown in this thesis, by passivating the lower side of the slab you can converge the electronic structure at the surface with ~4 rather than with 9 Au layers - not bad!
Unfortunately, however, that's not where complications end: a perfect Au(111) surface does not exist in nature.
Instead, the breaking of bonds at the surface causes the Au(111) surface to reconstruct, which increases the size of the surface unit cell from $1\times 1$ to $22\times\sqrt{3}$.
Finally, if you're creating your surface by polishing a crystal, you're going to have (reconstructed) Au(111) terraces of finite width as shown in the figure below (Figure 3.13 from Ph.D. thesis of Roberto Gaspari).
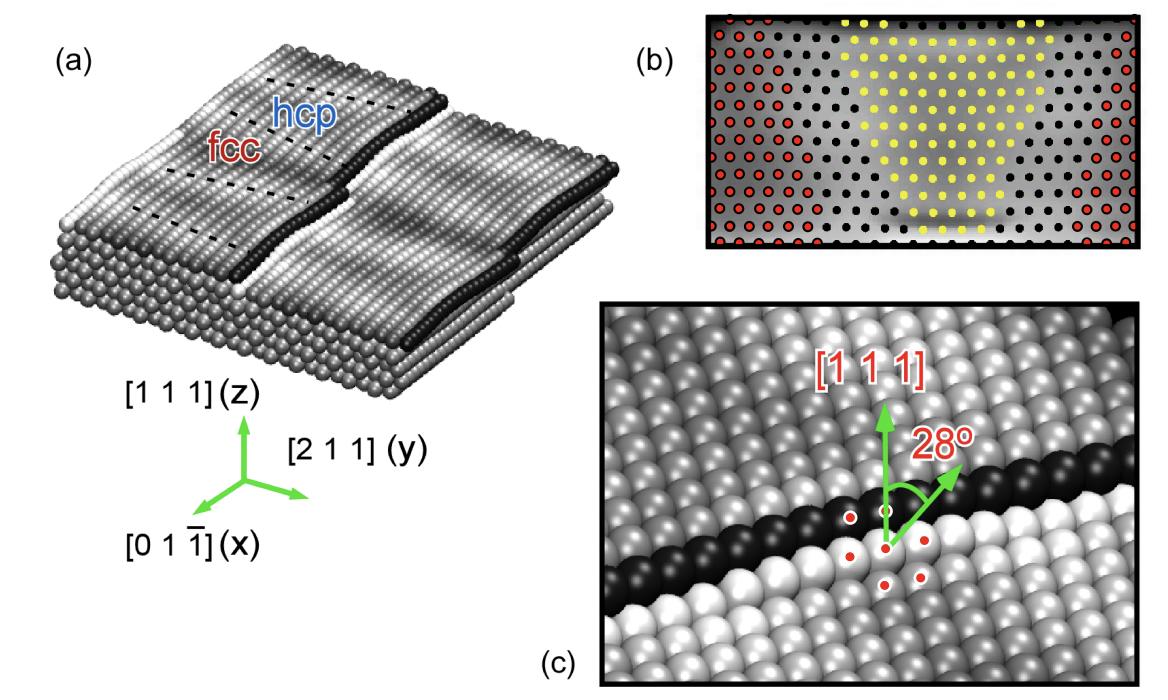
Of course, the steps in between terraces have their own peculiar electronic structure - at some point, the question always becomes: how accurately do you need to represent reality?
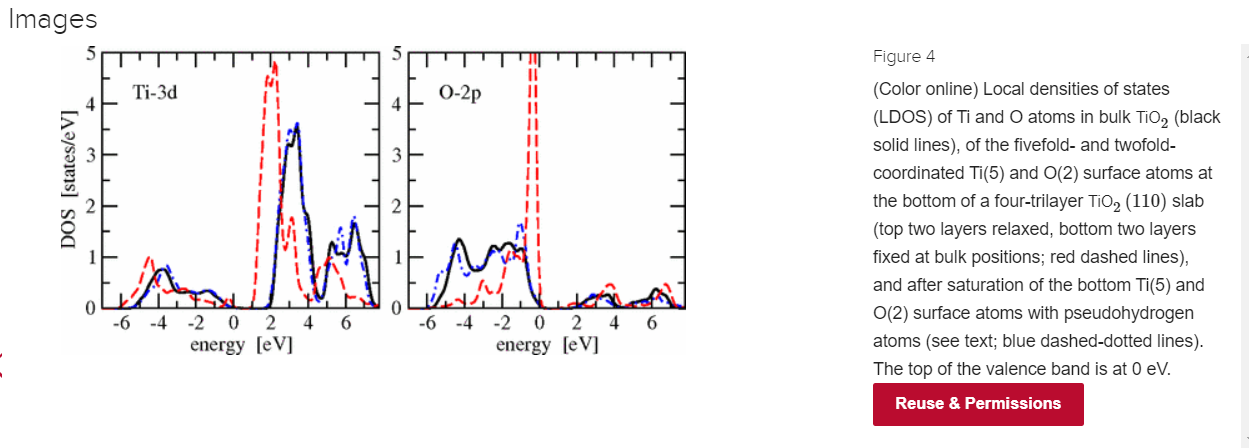
I'm not really familiar with the TiO2(110) surface but I read in chapter 3.5 of this thesis by Anders Rønnau that this termination involves substantial atomic relaxation of the subsurface layers, so it seems reasonable that a suitable passivation will help.
1 One aspect we don't discuss here are polar surfaces (surfaces that contain a net charge). For such cases, a dipole correction will be necessary for the correct description of the electric field at the slab's surface.
2 For a more rigorous analysis of when surface states arise in the tight binding picture, see e.g. here. Amongst other conclusions, Forstmann mentions "On surfaces of threedimensional crystals, one expects as many surface states as there are surface atoms" - but it is important to understand the assumptions that go into this statement (it is certainly not true in general).