For background, I have never used VASP before and have limited knowledge of how the software works. I am working on a program for the automation of structural creation for 2D materials. I am more of a computer scientist then a computational chemist and someone else is employing all my programs for their VASP optimizations and band width calculations.
Currently, my program takes in coordinates for metal centers, organic linkers, etc... and, along with some other user defined parameters, writes a .xyz file for the desired periodic structure. The coordinates from my program are extracted and another program (that I did not write) uses them to write a POSCAR file. However, when the periodic structure is visualized in VESTA with the unit cell vertices, the structure is not centered in the unit cell. At this point, I was asked to make a series of geometrical transformations to the original file in order to get the structure to the desired location.
I proposed that we adjust the lattice vectors/parameters to center it based off of the periodic structure coordinates as opposed to using the same lattice parameters and transforming each structure individually, as the former might be more efficient. However, I was told that this is not possible.
Here is my question
Is it possible to define the vertices of the unit cell around the structural coordinates?
If the situation or question is unclear, I can try and explain it better with diagrams or pictures, however, I apologize in advance as my knowledge in this subject is very limited.
EDIT:
I did not make these in VASP or VESTA but just made some pictures in Microsoft word to represent the situation as I don't have access to my lab computer right now. Hopefully this makes the question more clear.
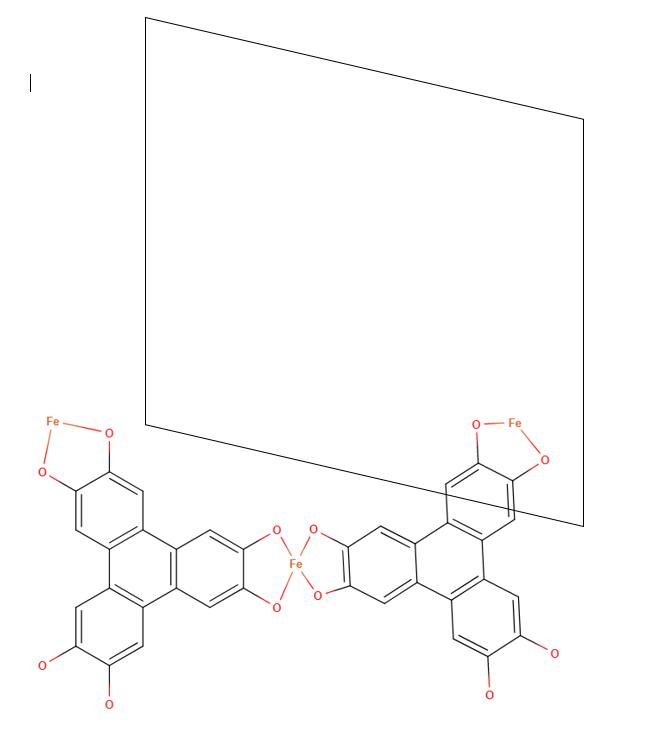
It initially looks like this when the structure and unit cell vertices are rendered in VESTA. Then, after essentially doing guess and check transformations with the original .xyz file, the structure is transformed to look like this.
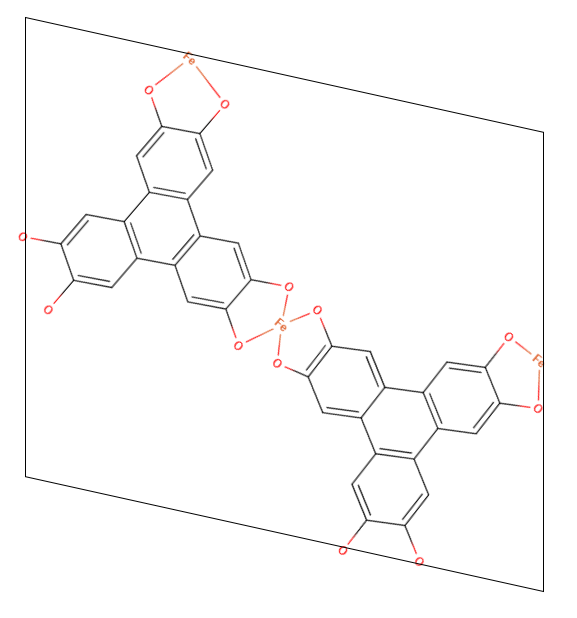
So instead of transforming the initial structure, is it possible to change the origin of the basis set for the unit cell vertices (i.e. move the box down and center it based off of the initial molecular coordinates provide.