"Inversion" is common in wavefunction-based quantum mechanics, for example the RKR inversion method which constructs a potential energy function based on information that can be obtained from spectroscopic experiments, such that a Hamiltonian using this potential, when fed through the Schroedinger equation, will give eigenvalue differences that match the experimentally observed spectra. Likewise similar things can be done if the experimental data doesn't only contain energies (eigenvalues) but contains wavefunction information (such as Franck-Condon factors or spectroscopic intensities). The RKR method has a lot of historical significance, and it still sometimes used today, including (sometimes) as a stepping stone towards "Direct Potential Fitting" which is the state-of-the-art method for obtaining an empirical potential function for a small molecule from experimental spectroscopic data.
A similar thing could be done if we had an "experimental" ground-state electron density, because:
HK Theorem 1: The external potential, is a unique functional of the electron density
which means $V(\mathbf{r}_i) = V[n(\mathbf{r})]$, which can then be plugged into the canonical expression for the many-electron time-dependent Hamiltonian:
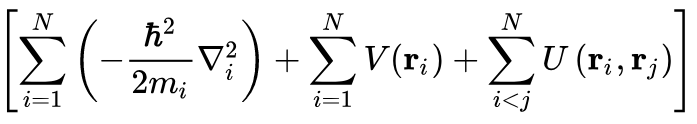
Then since the kinetic energy (the first sum) and the electron-electron interaction energy (third sum) are "universal operators", meaning that they're the same for any $N$-electron system, plugging in our obtained $V(\mathbf{r}_i)$ gives us a completely characterized Hamiltonian, which means all properties of the system, including the many-body wavefunction follow, though if we had the density we could have just got those properties properties from:
$$\left\langle \hat{O} \right\rangle = \textrm{Tr} \left(\hat{\rho}\hat{O} \right), ~ \hat{\rho} = n(\mathbf{r}).
\tag{2}$$
There's some caveats:
Just as in the "Direct Potential Fitting (DPF)" using wavefunction-based quantum mechanics where you need a "model" for the potential energy function, if you don't know the functional $V[n(\mathbf{r})]$ you would need to use a model functional and the accuracy of the $V(\mathbf{r})$ that you get, will depend on how good your model functional is.
Obtaining the Hamiltonian can be considered obtaining the "material's properties" (to use your words), but by "material properties" you might mean things like "conductivity". In the DPF case we do obtain things like bond lengths from the fitting procedure, but only because bond length is a property defined based on an easily accessible property of the potential energy function which was obtained in the DPF process: likewise any properties of the material which are defined based on easily accessible properties of $V(\mathbf{r})$, will be obtained, but other properties which are defined differently, could be better obtained from Eq. 2.
Finally to answer your follow-up question:
"As a followup, what experimental accuracy for ρ(r) is needed to accurately determine those material properties?"
You can use error propagation techniques to determine what an error in $n(\mathbf{r})$ will imply for the errors of $\left\langle \hat{O} \right\rangle$ from Eq. 2, but for obtaining properties based on $V(\mathbf{r})$ this paper by Bob Le Roy would be a good place to start, but beware of the fact that there will be a contribution to the final uncertainty in the material property coming from the choice of the model (functional in your case, potential energy functioni n the Schroedinger-based DPF case) you use, which can be extremely difficult even to define, and is unfortunately ignored usually in research papers even by experts.